10_DE_analysis
By Yan Li
PhD in Bioinformatics, University of Liverpool
RNA-seq
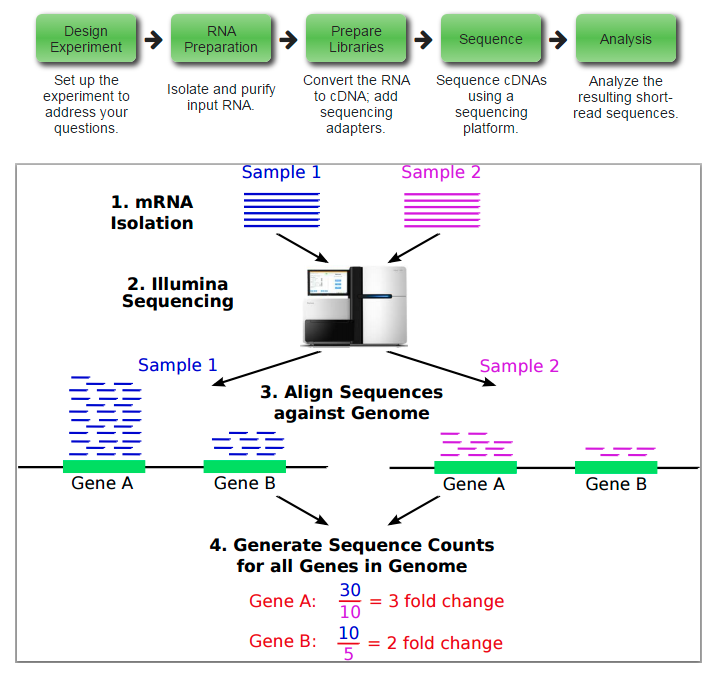
Popular software
- Mapping:
Tophat2,HISAT2 - Reads counting:
HTSeq-counts,Cufflink - Defferential Expressing analysis:
EdgeR,DEseq2,limma - GO enrichment:
DAVID,g.profiler
Terminology
- RPKM: Reads Per Kilobase per Million mapped read.
- RPKM = 10^9 * N / L * 1 / C
- N: the total number of reads mapped to a transcript
- C: the number of reads mapped to a gene
- L: the length of the gene
- GO: Gene ontology
Basic Statistics
- P-value and False discovery rate (FDR) adjusted p values
- a p-value of 0.05 implies that 5% of all tests will result in false positives.
- An FDR adjusted p-value (or q-value) will result in fewer false positives.
- Fold Change
- Fold change is a measure describing how much a quantity changes going from an initial to a final value.