Coverage / Depth
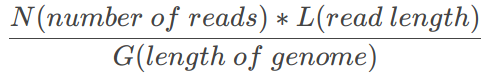
- Usually expressed as 30x, 100x, etc
- Low coverage cause some genome regions have no reads
- Short reads length may make repeat regions impossible to recover
\frac{N(number\space of\space reads) * L (read\space length)}
https:://katex.org/#demo
Graph theory: the Seven Bridges of Königsberg
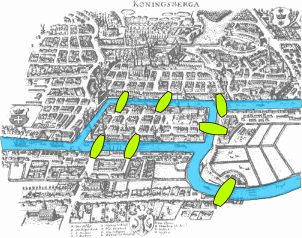
Can we visit each part of the city by crossing each bridge once?
Graph theory: the Seven Bridges of Königsberg

- Eulerian path = visit every edge of the graph only once
- In this problem it's impossible
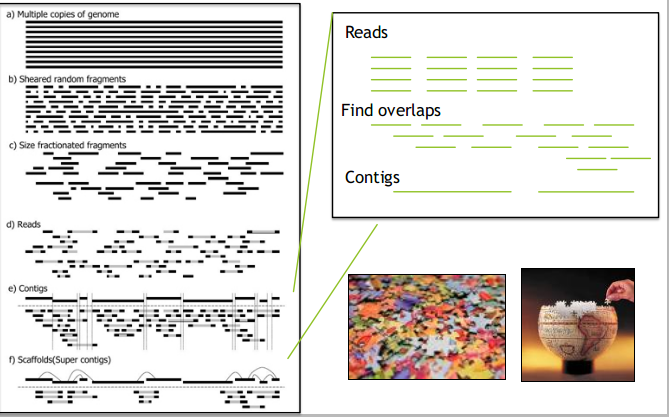
de Bruijn graph assembler
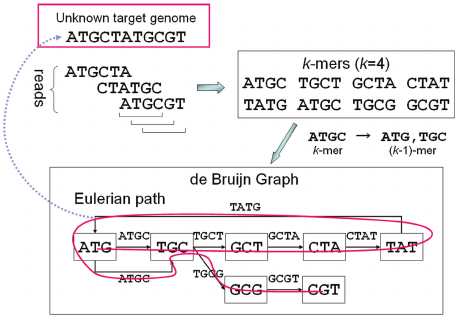
Graph features
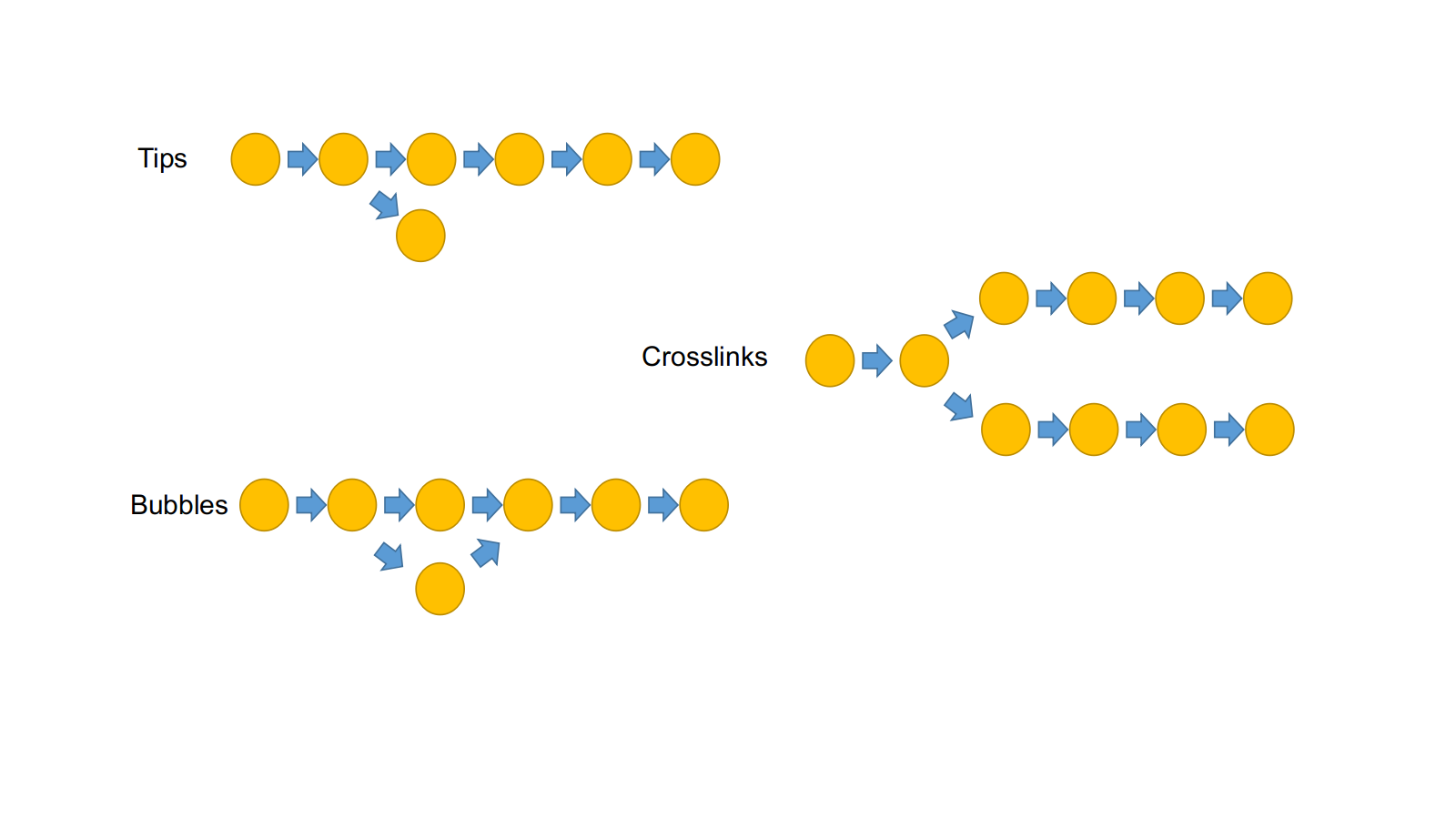
Use k-mer frequency to resolve these graph features::
- remove low depth kmers
- clip tips, merge bubbles, remove links
- resolve small repeats using long kmers